About Project
Objectives
Application



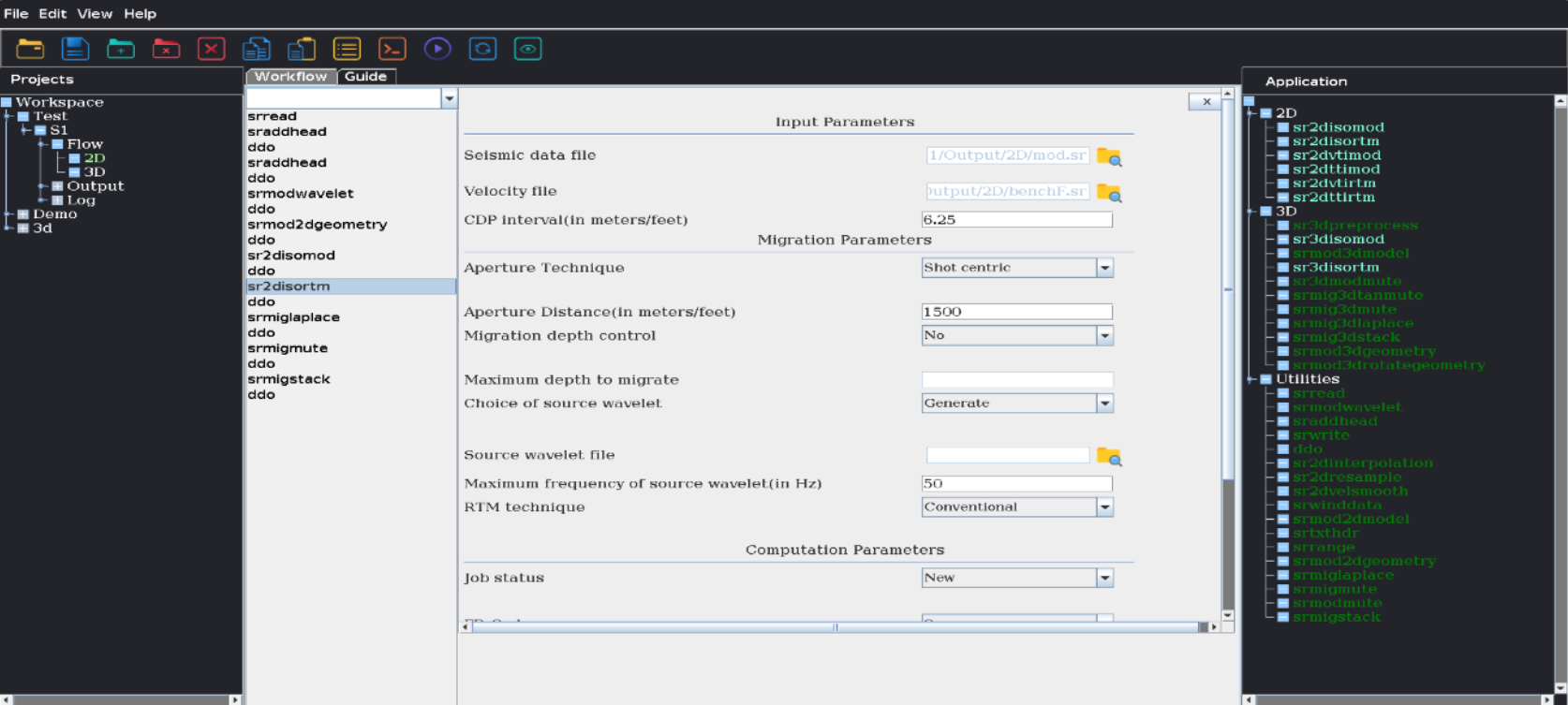
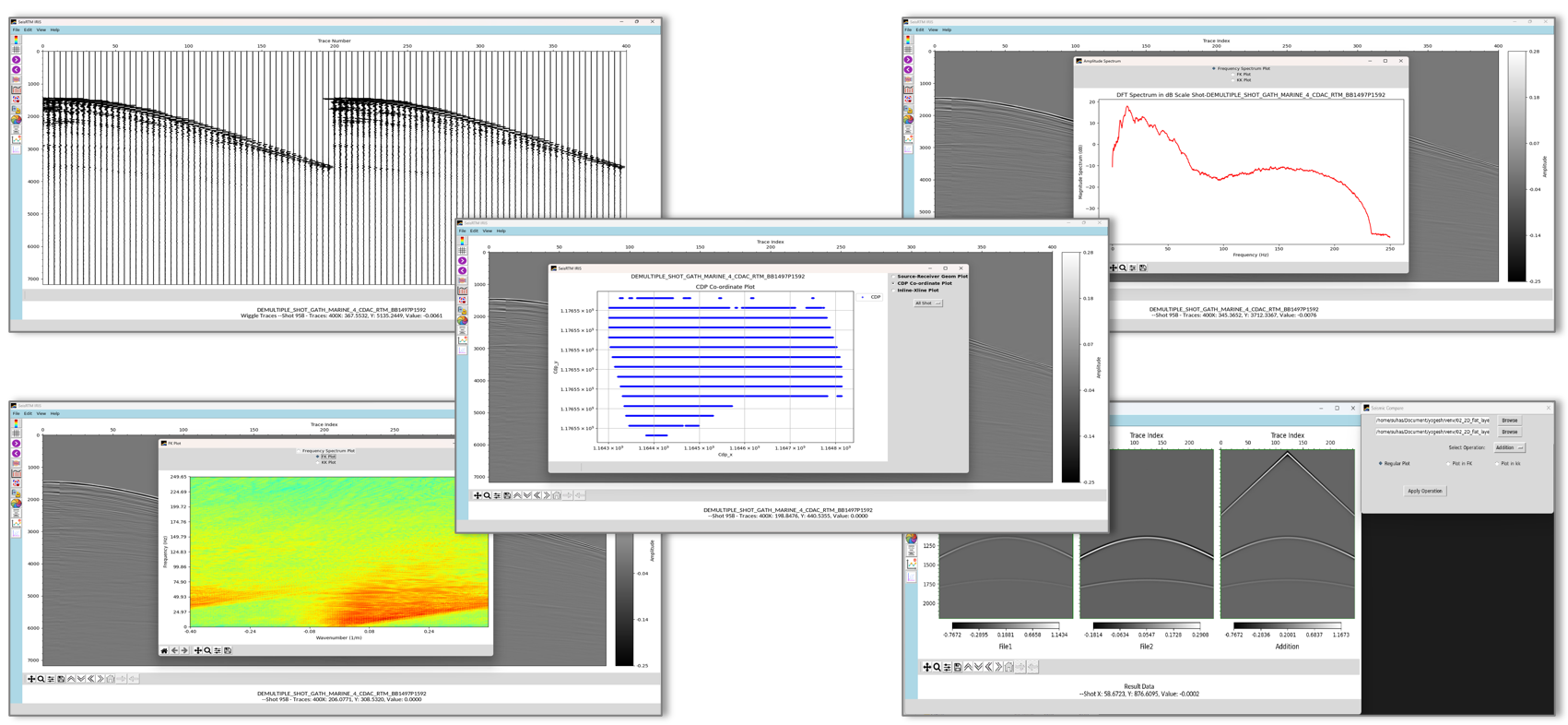
Seismic Data is acquired on land and in marine environment to find out the location of oil & gas reservoir in the subsurface. The processing of seismic data involves several steps before it gets converted into a meaningful and interpretable image of earth volume. Seismic Imaging is an advance processing step which converts seismic data to an earth subsurface image. Seismic imaging aims to create high resolution 2D & 3D structural images of subsurface geology. Reverse Time Migration (RTM) is one of the most reliable and preferred solution for seismic imaging of geological subsurface with complex structures. It can handle large velocity variation without any dip limitations for producing the earth subsurface structure with high resolution. It can deliver subsurface images with high accuracy in different mediums. RTM is highly computation, I/O and storage intensive, which requires High Performance Computing (HPC) ecosystem for execution.

Under the National Supercomputing Mission (NSM), “A HPC software suite for seismic imaging to aid oil and gas exploration” is a “Make in India” initiative to develop a customizable and efficient RTM software “SeisRTM”. It can provide high-resolution 2D & 3D seismic images of complex geological subsurface using acquired large seismic data. This software provides 2D modelling and RTM capabilities for both isotropic and anisotropic (VTI and TTI) media, as well as 3D isotropic modelling and RTM. SeisRTM is equipped with a suite of data preparation and post-processing tools, optimized to handle high-frequency migration and large datasets efficiently. It is designed for parallel computing environments, making it well-suited for deployment on CPU clusters without core limitations. SeisRTM offers both a Command Line Interface (CLI) and a Graphical User Interface (GUI) for user flexibility and includes an in-house developed data visualization tool to streamline data analysis and visualization. The indigenously developed “SeisRTM,” built on NSM infrastructure, will serve as a seismic imaging facility, delivering 3D RTM capabilities for upstream oil and gas exploration companies in India.

A Seismic Imaging Solution






Centre for Development of Advanced Computing (C-DAC), Pune
Geodata Processing and Interpretation Centre (GEOPIC), ONGC
Indian Institute of Technology Roorkee (IITR)













Scientific research is pursued through the combination of three essential practices: theory, experiment, and computation. Since the emergence of modern computers, computation for modelling and simulation of experiments has led to the emergence of Computational Science. High-performance computing (HPC) has a major role to play to process data and perform complex calculations at high speeds. HPC applications are the drivers of the development of both hardware and software for HPC systems. Computations on HPC for solving many real-life scenarios to solve complex problems in science, business, and engineering have immense potential to influence the economy and the quality of life for human kind. Materials science is a field involving research and discovery of materials. Computational materials science uses modeling, simulation, theory, and informatics to understand materials. The main goals include discovering new materials, determining material behavior and mechanisms, explaining experiments, and exploring materials theories. It is one sub-discipline of both computational science and engineering, containing significant overlap with computational chemistry and computational physics
The outcome of the project “Materials and Computational Chemistry” under National Supercomputing Mission (NSM) is the set of codes (software) developed by the investigators to perform the computations to study properties of atoms, molecules, clusters, alloys, bio-molecules, and composite materials using high-performance computing (HPC).
Development of indigenous scientific codes:(l)Linear Scaling DFT,(2)Multi-Reference Methods with hybrid QM-MM approaches,(3)Excited state dynamics toolkit, (4) Multiscale Microstructure Simulation and Modelling, (5) GUI for home-grown quantum chemistry code
Development of indigenous scientific codes
*Since March 2023, Materials and Computational Chemistry application support activity was added [on the reccomendations of the PMC and TAC] for the proliferation of the five indigenous software developed.
Five indigenously developed software in the domain of Materials and Computational Chemistry, Publications in reputed international journals, One training workshop for all five software.
Five indigenously developed and open-source software:
31 Publications in International Journals from five sub-projects
Efforts for the proliferation of the software among the user community


Seismic Data is acquired on land and in marine environment to find out the location of oil & gas reservoir in the subsurface. The processing of seismic data involves several steps before it gets converted into a meaningful and interpretable image of earth volume. Seismic Imaging is an advance processing step which converts seismic data to an earth subsurface image. Seismic imaging aims to create high resolution 2D & 3D structural images of subsurface geology. Reverse Time Migration (RTM) is one of the most reliable and preferred solution for seismic imaging of geological subsurface with complex structures. It can handle large velocity variation without any dip limitations for producing the earth subsurface structure with high resolution. It can deliver subsurface images with high accuracy in different mediums. RTM is highly computation, I/O and storage intensive, which requires High Performance Computing (HPC) ecosystem for execution.
Under the National Supercomputing Mission (NSM), “A HPC software suite for seismic imaging to aid oil and gas exploration” is a “Make in India” initiative to develop a customizable and efficient RTM software “SeisRTM”. It can provide high-resolution 2D & 3D seismic images of complex geological subsurface using acquired large seismic data. This software provides 2D modelling and RTM capabilities for both isotropic and anisotropic (VTI and TTI) media, as well as 3D isotropic modelling and RTM. SeisRTM is equipped with a suite of data preparation and post-processing tools, optimized to handle high-frequency migration and large datasets efficiently. It is designed for parallel computing environments, making it well-suited for deployment on CPU clusters without core limitations. SeisRTM offers both a Command Line Interface (CLI) and a Graphical User Interface (GUI) for user flexibility and includes an in-house developed data visualization tool to streamline data analysis and visualization. The indigenously developed “SeisRTM,” built on NSM infrastructure, will serve as a seismic imaging facility, delivering 3D RTM capabilities for upstream oil and gas exploration companies in India.
Centre for Development of Advanced Computing (C-DAC), Pune
Geodata Processing and Interpretation Centre (GEOPIC), ONGC
Indian Institute of Technology Roorkee (IITR)
UES2S is an online fully coupled urban ‘meteorology, hydrology, and air quality’ modeling system developed under the NSM Urban Modelling Project (Figure 1). This system captures the urban representation of micro-scale city environmental conditions. UES2S has three major components: Data as a service (DataHub), modeling platform as a service (Science Gateway), and Decision Support System (DSS) for cross-sector end-user decisions. Through DataHub, we intend to provide cross-sector data access and a data-sharing facility. The Science Gateway (Figures 2 & 3) has automatic end-end modeling workflows enabling ready-to-use weather, hydrology, and air quality models on NSM clusters. The DSS component (Figures 4 & 5) facilitates the translation of scientific data into multi-stakeholder interactive actions. The DSS provides high-resolution weather, air quality, and hydrology forecasts along with the forecast of reservoir inflows, water levels, and discharge for flood management and mitigation. Thus, the DSS is integral to disaster management activities, daily operations, and science-based policy decisions.

This multi-sectorial simulation lab and science-based decision framework is developed to address urban environment issues. This HPC-based automated model execution workflows with an interdisciplinary urban testbed has been developed to execute weather, air quality, and hydrology models for the prediction of extreme events. The system is designed to be exceptionally user-friendly, enabling researchers and students to execute the models easily. This would facilitate the seamless transition of research into operational practices. The framework offers an urban modeling system, operational processes, a data hub, and a DSS, enabling meteorology, air quality, and hydrology services for diversified user categories.
Science Gateway (Figure 2) is a digital platform customized for meteorologists, hydrologists, and air quality modelers, offering convenient access to data from specialized tools and collaboration features to enhance research and forecasting in these areas.
Workflow of the Weather Research and Forecasting (WRF) (Figure 3) is developed in Science Gateway, a state-of-the-art mesoscale numerical weather prediction system designed for atmospheric research and operational forecasting applications.
A Decision Support System (DSS) (Figure 4) helps in decision-making during extreme events like heavy rainfall, floods, and heatwaves. It facilitates analyses of meteorological patterns like short-duration high-intensity rainfall, heat and cold waves, hydrological parameters such as reservoir levels and river flows, and air quality parameters like PM 2.5. For instance, during an extreme event, the DSS provided accurate forecasts and risk assessments through charts, shaded and non-shaded plots, and vector plots, assisting users in rainfall information, flood control, air quality monitoring, and climate resilient planning.
The reservoir operations module, which is a key component of the DSS, displays a time series forecast plots as shown in Figure 4. This plot includes various parameters such as upstream/downstream catchment rainfall, reservoir water level, dam discharge, and reservoir inflow for the user-selected reservoir. The user, typically a scientists or operational forecasters in weather, environmental science, or disaster management, can use this information to make informed decisions and plan accordingly.
In one of the DSS module (Figure 5), users can select flood hotspots in the alerts section and then click on a hotspot pin for a particular location to visualize water depth relative to human height, making the representation more intuitive and engaging.






